تکنیکهای نوین توالییابی با کارایی بالا به عنوان رهیافت نوینی در شناسایی واریانتهای ژنتیکی و اطلاعات عملکردی در گونههای بسیاری از چمانگان قرار گرفتهاند. اسب کاسپین با توجه به ویژگیهای منحصر به فرد ژنتیکی و فنوتیپی خود، یکی از نژادهای مهم اسب در ایران و جهان است.
از اینرو، پژوهش حاضر به شناسایی واریانتهای ژنتیکی چندشکلیهای تک نوکلیوتیدی، حذف و اضافههای کوتاه و واریانتهای تعداد در نسخه (CNV) در اسب کاسپین و بررسی نقش آنها در فرآیندها و مسیرهای بیولوژیک ویژه پرداخته است. با استفاده از توالییابی با کارایی بالا، Gb108 از DNA ژنومی سه مادیان کاسپین با میانگین عمق 8/45 توالییابی شدند. این توالییابی با میانگین همپوشانی 41/14 کم و بیش 4/76 درصد از ژنوم رفرانس اسب را پوشش داد. با استفاده از فیلترینگ سختگیرانه 1666717 چندشکلی تک نوکلیوتیدی، 358020 حذف و اضافههای کوتاه و CNV 3109 شناسایی شدند. کلاسترینگ عملکردی واریانتهای ژنی اسب کاسپین نشان داد که بیشتر این واریانتها در ژنهای مرتبط با فرآیندهای سیستم عصبی، تنظیم ورارسانی فرستههای بیوشیمیایی مرتبط با گوانیدین تریفسفات، موفوژنز سلولی، سازمانبندی اسکلت سلولی، توسعه رگی، جنبایی سلولی و انتقال غشایی روی داده است. فزون بر این، واریانتهای ساختاری ژنوم اسب کاسپین مانند اینورژنها و جابهجا شدگیها و نیز حذف و اضافههای بزرگ ژنومی که در این پژوهش شناسایی شدند، میتوانند در طراحی نشانگرهای ژنتیکی برای کارهای اصلاح نژادی و نیز بررسیهای جمعیتی سودمند واقع شوند.
واژههای کلیدی: توالییابی با کارایی بالا، اسب کاسپین، واریانت ژنتیکی، مسیرهای بیولوژیک.
نویسندگان:
بابک عارف نژاد1، حمید کهرام2، محمد مرادی شهربابک3، ملک شاکری2، یانگ دونگ5، خیائولی ژانگ5، ون وانگ5، قاسم حسینی سالکده*4
1-دانشجوی دکتری فیزیولوژی دام –گروه علوم دامی-پردیس کرج-دانشگاه تهران
2-استادیار گروه علوم دامی، پردیس کرج-دانشگاه تهران
3-دانشیار گروه علوم دامی، پردیس کرج-دانشگاه تهران
4-استادیار گروه ژنومیکس پژوهشکده بیوتکنولوژی کشاورزی-کرج
5- انستیتو حیات وحش کانمینگ-آکادمی علوم چین
مقدمه
اسب کاسپین یکی از کهنترین نژادهای اسب در دنیا است که خاستگاه آن به بیش از 3000 سال پیش بر میگردد. گفته میشود که این نژاد جد اولیه همه اسبهای خونگرم دنیا است و همانندی زیادی با اسبهای عرب دارد. اسبهای کاسپین نخستین بار در سال 1969در شمال ایران شناخته و معرفی شدند (Firouz, 1969). لوییس فیروز پس از کشف دوباره این اسبها ویژگیهای مورفولوژیک آنها را نزدیک به اسبهای نگارههای پرسپولیس شرح داد. این اسبها به غیر از ارتفاع بدن، همانند دیگر اسبها هستند و تفاوتهای آناتومیک ناچیزی با دیگر اسبها دارند (Firouz, 1971; Firouz, 1972). بنابراین، این نژاد به علت کوتاهی قد به فرنام پونی[1] نیز شناخته شده میشود ولی در برابری با دیگر نژادهای پونی در جهان که اسبانی با بدنی چاق و کوتاه و اندامهای حرکتی قوی و زمخت و بدون تناسب اندام هستند، بیدرنگ این اسب از این نوع نژاد مجزا شده و به اسب مینیاتوری معروف شده است (Firouz, 1972).
نرخ آبستنی در این اسبها کمتر از 40 درصد است و گفته میشود که از مهمترین مشکلات تولیدمثلی آنها نرخ تخمکریزی پایین در آنها است. در نریانها نیز شمار اسپرم و جنبایی اسپرم پایین است که بازدهی پایین تولیدمثلی را در این نژاد نشان میدهد (Hatami-Monazah and Pandit, 1979). به علت اندازه کم جمعیت در این اسبها، تنوع ژنتیکی در این توده پایین است که این امر موجب کاهش شایستگی[2] و افزایش همخونی[3] در آنها شده است. از اینرو، این اسبها در دسته حیوانات در حال انقراض قرار میگیرند. بنابراین، سازوکارهای شناسایی گوناگونی ژنومیک (به ویژه در ژنهای تولیدمثلی و نیز ژنهای مرتبط با سازگاری) در بهبود شناخت بیشتر در سطح مولکولی و نیز به کارگیری روشهای اصلاح مولکولی اهمیت ویژهای در این نژاد دارند. (Shahsavarani and Rahimi-Mianji, 2012).
در دسترس بودن توالیهای ژنومی دریچهای بر تعیین ژنوتیپ با کارایی بالا و در مقیاس گسترده گشوده است. در آغاز، تکنولوژیهای تعیین ژنوتیپ بر پایه ریزآرایههای DNA (که توانایی شناسایی SNPها را در سطح ژنوم دارند) گسترش یافتند. برکسی پوشیده نیست که این روشهای تعیین ژنوتیپ، بازدهی شناسایی هزاران نشانگر را در یک فرآیند هیبریدسازی DNA ژنومیک با الیگونوکلیوتیدهای قرارگرفته شده روی Gene Chip بهبود بخشیدهاند (Huang et al., 2009; Winzeler et al., 1998). هرچند که با این تکنیک، کم و بیش هدف بررسی نشانگرها در مقیاس گسترده محقق شده است، ولی همچنان روشهای بر پایه ریزآرایه DNA دارای محدودیتهای جدیای هستند. برای نمونه، طراحی، تولید و نیز همه فرآیند شناسایی نشانگرها با این تکنیک دشوار، زمانبر و پرهزینه است (Huang et al., 2009).
نسل جدید تکنولوژیهای توالییابی همراه با توالیهای در دسترس بیشماری از ژنومهای گوناگون، دستیافت تازهای را برای طراحی دوباره استراتژیهای تعیین ژنوتیپ، نقشهیابی ژنتیکی و نیز آنالیزهای ژنومی ارایه کرده است. تکنیکهای جدید توالییابی نه تنها کارایی و همپوشانی توالییابی را به گونه چشمگیری افزایش دادهاند، بلکه امکان توالییابی شمار زیادی از نمونههای زیستی را با استراتژی توالییابی Multiplex فراهم کردهاند (Craig et al., 2008; Cronn et al., 2008; Huang et al., 2009). روی هم رفته، این تکنیکها در گسترش روشهای تعیین ژنوتیپ در مقیاس گسترده و بر پایه توالییابی ژنوم پیشرفتهای روزافزونی داشتهاند به گونهای که همپوشانی، صحت و دقت در نقشهیابی بسیار چشمگیر است و مقایسههای ژنوم و نقشههای ژنومی در میان ارگانیسمها و جمعیتهای گوناگون سنجشپذیرتر است. از این رو، این تکنیکها فزون بر اینکه آنالیزهای ژنتیک با هدف شناسایی واریانتها در مقیاس گسترده را به گونهای چشمگیر آسان و کارآمدتر میکنند، پاسخهای دقیقتری به پرسشهای بیولوژیک ارایه میکنند (Cronn et al., 2008; Huang et al., 2009).
در جمعیتهای گوناگون یوکاریوتها، چندشکلیهای تکنوکلیوتیدی (SNP)[4] و نیز واریانتهای CNV[5] در ژنوم از منابع مهم واریاسیونهای ژنتیکی و فنوتیپی هستند. فزون بر این، چندشکلیهای INDEL[6] نیز کم و بیش فراوانند و در بروز صفات و فنوتیپهای گوناگون کنشهای معنیداری دارند. با این وجود، به علت دشواری و بازدهی پایین آنها در پلاتفورمهای بر پایه تکنیکهای ریزآرایه، تاکنون کمتر شناسایی و بررسی شدهاند؛ ولی امکان بررسی همه جانبه و نیز نقشهیابی همه اینگونه از واریانتها با کمک تکنیکهای جدید توالییابی و تکامل دانش نوظهور بیوانفورماتیک فراهم شده است (Shao et al., 2012).
تاکنون ژنومهای اسبهای Quarter (Doan et al., 2012)، اسب عرب، Icelandic، Standardbred، Norwegian_Fjord، Thoroughbred، Przewalski (Orlando et al., 2013) با استفاده از این تکنیک توالییابی شدهاند. با وجود اهمیت اسبهای ایران به ویژه اسب کاسپین در ذخایر ژنتیکی اسب هیچگونه پژوهشی روی ساختار ژنوم و واریانتهای ژنومی آنها انجام نشده بود. از این رو، پروژه حاضر آغازی بر اینگونه بررسیها در اسبهای ایرانی است. هدف اصلی این پژوهش، تعیین ساختار ژنوم و نیز شناسایی واریانتهای رایج در ژنوم اسب کاسپین و شناسایی مسیرهای بیولوژیک مرتبط با آنها با کمک تکنیکهای نوین توالییابی بود.
مواد و روشها
نمونهگیری و استخراج DNA از سلولهای سفید خون
سه نمونه خون از مادیانهای کاسپین در موسسه تحقیقات خجیر (پارک ملی خجیر-تهران) گرفته شدند. نمونهها در فلاسک یخ به آزمایشگاه منتقل و بیدرنگ پس از جداسازی سلولهای سفید از قرمز خون، DNA با روش استخراج نمکی تغییر یافته استخراج و کیفیت و کمیت آن با کمک اسپکتروفتومتری و الکتروفورز با ژل آگاروز تعیین شد. برای ساختLibrary DNA و تعیین توالی به انستیتوی BGI[7]
(BGI, Shenzen, China) در چین فرستاده شدند. توالییابی ژنوم اسب کاسپین با استفاده از پلاتفورم Hiseq2500 Illumina با اندازه bp300 و توالییابی دو سویه انجام شد و دادههای خام ژنومی که از توالییابی به دست آمدند برای آنالیزهای بیوانفورماتیک پردازش شدند.
پیشپردازش توالیهای کوتاه ژنومی
کنترل کیفی توالیهای کوتاه ژنومی به دست آمده از توالییابی اسب کاسپین با استفاده از Fastqc (Andrews, 2012) انجام شد. پس از آن، توالیهای کوتاه دو سویه ژنومی با استفاده از AdapterRemoval v1.2 (Lindgreen, 2012) به سه دسته توالی منفرد، کولاپس شده و دو سویه پردازش شدند. در این فرآیند، همزمان با حذف بخشهایی از توالی که کیفیت خوانش پایینی داشتند توالیهای دوسویهای که دستکم در bp11 با هم همپوشانی داشتند، کولاپس شدند و به عنوان یک توالی منفرد درنظر گرفته شدند. در بخشهای کولاپس شده توالیهای کوتاه، Phred Quality Score و نوکلیوتید مربوطه بر اساس بالاترین اسکور نگهداشته شدند. در مواردی که هیچ همپوشانی میان دو سوی خوانش توالیهای کوتاه پیدا نشد، توالی جداگانه پردازش و بخشهای با کیفیت خوانش پایین، نوکلیوتیدهای N و نیز توالیهای آداپتوری از آنها حذف شدند. پس از این فرآیند، توالیهایی که کمتر از 25 نوکلیوتید طول داشتند حذف شدند.
همردیفی دادههای به دست آمده از ژنوم اسب کاسپین
پس از پالایش توالیهای کوتاه ژنوم اسب کاسپین، فرآیند نقشهیابی آنها با 31 کروموزوم اتوزوم و کروموزوم X ژنوم رفرانس اسب (http://genome.ucsc.edu) انجام شد. در این فرآیند، با استفاده از BWA 0.5.9[8] (Langmead, 2002) پس از ایندکس سازی ژنوم رفرانس، همه توالیهای کوتاه با کمک این الگوریتم و تعیین پارامترهای مناسب با ژنوم ایندکس شده، همردیف شدند. توالیهای منفرد و کولاپس شده با استفاده از bwa samse و توالیهای کوتاه دو سویه با استفاده از bwa sampe همردیف شدند. پس از آن توالیهای کوتاه مضاعف شده در PCR با استفاده از MarkDuplicates نرمافزار Picard tools نسخه 1.99 (http://picard.sourceforge.net) حذف شدند. در پایان فایلهای BAM که برای هر دسته از توالیهای کوتاه به دست آمدند با استفاده از MergeSam با یکدیگر ادغام شدند. تک فایل BAM به دست آمده برای افزایش صحت همردیفی و نیز شناسایی واریانتها، با استفاده از الگوریتم GATK [9] (McKenna et al., 2010) و بر اساس INDELهای شناخته شده اسب دوباره همردیف شدند.
شناسایی واریانتهای ژنوم اسب کاسپین
در این فرآیند، پس از کالیبراسیون دوباره کیفیت خوانش توالیهای کوتاه ژنومی بر اساس کوواریتهای Read Group، Quality Score، Cycle و Dinucleotideها برای تک فایل BAM به دست آمده از گامههای پیشین، واریانتهای SNP با استفاده از الگوریتم GATK UnifiedGenotyper به کمک Queue و اسکریپت Scala شناسایی شدند. پس از شناسایی واریانتها، SNPها با استفاده از فیلترینگ سختگیرانه GATK برای QualByDepth کمتر از 2، FisherStrand بیشتر از 60، RMSMappingQuality کمتر از 40، HaplotypeScore بیشتر از 13، MappingQualityRankSumTest کمتر از 5/12 و ReadPosRankSumTest کمتر از 8 فیلتر شدند. INDEL واریانتهای INDEL نیز با استفاده از Freebayes v-0.9.9 با الگوریتم بیزین در شناسایی واریانتها استفاده شد (Garrison and Marth, 2012) و پس از آن برای QualByDepth کمتر از 2، FisherStrand بیشتر از 200 و ReadPosRankSumTest کمتر از 20 فیلتر شدند. واریانتهای CNV و INDELهای بزرگ نیز با استفاده از الگوریتم BreakDancer (Chen et al., 2009) و CNVnator (Abyzov et al., 2011) شناسایی و برای نواحی Gapهای ژنوم، تلومریک و سانترومریک فیلتر شدند.
شناسایی اثر واریانتها بر کنش ژنها و کلاسترینگ عملکردی
برای شناسایی اثر واریانتها بر کنش ژنها و نیز مکانیابی واریانت در بخشهای ساختاری ژنها از نرمافزار snpEff (Cingolani et al., 2012) و Annovar (Wang et al., 2010) استفاده شد. در این فرآیند، ابتدا واریانتهای به دست آمده از ژنوم اسب کاسپین با واریانتهای شناخته شده در دیگر نژادهای اسب مقایسه و پس از آن اثر واریانتها بر ژنها بررسی شدند. برای بررسی Gene ontology و کلاسترینگ عملکردی ژنهای دارای واریانتهای با اثر بالا بر کنش ژنها، از DAVID (Dennis Jr et al., 2003) استفاده شد.
نتایج
توالییابی ژنوم و پیشپردازش دادههای ژنومی اسب کاسپین
نتایج توالییابی، نقشهیابی توالیهای کوتاه و آنالیز قطعات ژنومی در جدول 1 نشان داده شده است. در فرآیند توالییابی روی هم رفته، 718859813 توالی کوتاه با 119 گیگا باز به دست آمد که پس از پالایش آنها بر اساس کیفیت توالیهای کوتاه به دست آمده از توالییابی مونتاژهای اتوزومی و کروموزوم X ژنوم رفرانس اسب (equCab2) نقشهیابی شدند. از 606810015 توالی کوتاه به دست آمده از فرآیند پالایش کیفیت (108 گیگا باز)، 368218734 توالی کوتاه (9/65 گیگاباز) با ژنوم اسب همردیف شدند و میانگین همپوشانی توالییابی ژنوم اسب کاسپین 41/14 و درصد همپوشانی با ژنوم رفرانس 4/68 محاسبه شد. شکل1 همپوشانی توالیهای کوتاه را در ژنوم اسب نشان میدهد.
شناسایی واریانتهای ژنتیکی
پس از همردیفی توالیهای کوتاه به دست آمده از توالییابی اسب کاسپین، واریانتهای SNP، INDEL و CNV آنالیز شدند. SNPها با معیارهای بسیار سختگیرانه الگوریتم GATK فیلتر شدند و کمترین میزان همپوشانی ژنومی برای شناسایی SNP، X5 در نظر گرفته شد. روی هم رفته، 1666717 چند شکلی تک نوکلیوتیدی (SNP) در ژنوم سه مادیان کاسپین در مقایسه با ژنوم رفرانس اسب شناسایی شد.
گوناگونی SNPها و INDELها در جدول 2 ارایه شدهاند. نرخ SNP برابر با 1 چندشکلی در هر 1418 باز بود. شمار Transition برابر 1155417 و شمار Transversion برابر 512986 بود که نسبت Transition/Transversion در اسب کاسپین 2523/2 برآورد شد. از مقایسه چند شکلیهای تک نوکلیوتیدی به دست آمده در این پژوهش با چند شکلیهای موجود در پایگاه اطلاعاتیSNP ((dbSNP, http://www.ncbi. nlm.nih.gov/projects/SNP نشان داد که 1448364 چند شکلی شناخته شده هستند و 218353 چند شکلی جدید هستند و تاکنون گزارش نشدهاند. شمار 1128216 چندشکلی تک نوکلیوتیدی در نواحی بین ژنی شناسایی شدند. چند شکلیهای شناسایی شده در فرادست و فرودست نواحی ژنی تا 5 کیلوباز فاصله با نواحی ژنی در نظر گرفته شدند. از همه چند شکلیهای ژنی شناسایی شده در اسب کاسپین 12910 چند شکلی نامعنی[10] و 18337 چند شکلی هممعنی[11] شناسایی شدند. فزون بر این، 73 چند شکلی بیمعنی[12] شناسایی شد. جایگزینیهای CCA/CCG با 546 مورد و GCT/GCC با 506 مورد بیشترین تغییرات کدون آمینواسیدی را در ژنهای اسب کاسپین به وجود آوردهاند. بیشترین شمار جایگزینیهای آمینواسیدی به آلانین/ترئونین (438)، آلانین/والین (348)، ایزولوسین/والین (304) و گلوتامین/آسپاراژین (354) اختصاص داشتند. از مجموع چند شکلیهای شناسایی شده در نواحی ژنی 361 چند شکلی با اثر تخریبی بالا، 12881 چند شکلی با اثر تخریبی متوسط و 18562 چند شکلی با اثر تخریبی کم در کنش ژن شناسایی شدند (جدول 2).
با در نظر گرفتن فیلترینگ سختگیرانه 358020 حذف و اضافههای (INDEL) کوتاه (کمتر از bp15) در ژنوم اسب کاسپین شناسایی شدند. روی هم رفته 211843 اضافههای نوکلیوتیدی و 146177 حذفهای نوکلیوتیدی شناسایی شدند. بررسی اثر این حذف و اضافههای نوکلیوتیدی بر کنش ژنها نشان داد که 4741 حذف و اضافه موجب تغییر الگوی نواحی رمزگردان ژنی می شوند در حالیکه 314 حذف و اضافه اثر متوسطی بر کنش ژن هدف دارند. فزون بر این، 13 حذف و اضافه موجب تغییر کدون امینواسیدی به کدون پایانی در نواحی رمزگردان ژنهای هدف خود میشوند (جدول2).
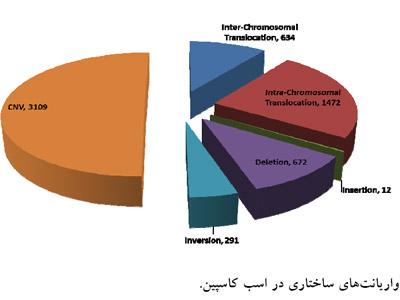
نتایج آزمون شناسایی CNV پس از تصحیح انحراف GC و فیلترینگ نواحی Gap ژنومی و تلومریک، شمار 3109 CNV را در ژنوم اسب کاسپین نشان داد که از این میان 902 کم شدگی (Loss) و 2207 زیاد شدگی (gain) شناسایی شدند. اندازه CNVها میان bp900 تا Mbp 86/2 بود. آنالیز ژنی CNVها نشان داد که 2307 CNV در نواحی بین ژنی و 802 CNV در نواحی ژنی و بیشتر در نواحی اینترونی قرار دارند. فزون بر این، 634 جابهجایی بین کروموزومی، 1472 جابهجایی درونکروموزومی، 672 حذفهای بزرگ، 12 اضافههای نوکلیوتیدی بزرگ و 291 اینورژن در ژنوم اسب کاسپین در مقایسه با ژنوم رفرانس اسب دیده شد (شکل2).
آنالیز کلاسترینگ عملکردی و Gene Ontology
آنالیز عملکردی و کلاسترینگ ژنهای دارای واریانتهای با اثر مخرب مسیرهای بیولوژیک مرتبط با واریانتهای ژنتیکی را نشان میدهد. هرچند اطلاعات مسیرهای بیولوژیک و Gene Ontology برای ژنهای اسب هنوز تکمیل نشدهاند. از این رو، با استفاده از اورتولوگهای انسانی مرتبط با ژنهای اسب، آنالیزهای عملکردی مرتبط با چتدشکلیهای تکنوکلیوتیدی بررسی شدند. از میان 5578 ژن دارای چند شکلی نامعنی، شمار 5665 ژن دارای اورتولوگ انسانی بودند و از این میان 5073 ژن دارای رکورد DAVID بودند. آنالیز کلاسترینگ نشان داد که چند شکلیهای نامعنی در اسب کاسپین با FDR[13] کمتر از 1 درصد در ژنهای مسیرهای بیولوژیک مرتبط با فرآیندهای سیستم عصبی، تنظیم ورارسانی سیگنال مرتبط با GTP، مورفوژنز سلولی، سازمانبندی اسکلت سلولی، توسعه رگی، جنبایی سلولی، سگنالینگ سلول-سلول، انتقال غشایی، فرآیندهای متابولیک RNAهای غیررمزگردان، تنظیم حرکت سلولی، تنظیم تولید سایتوکاینها، تشخیص محرکها، تنظیم ترشح، کاتابولیسم لیپیدها، تنظیم مثبت پاسخ به محرکها، فرآیندهای هومیوستاتیک و تکامل مغز پیشین نقش دارند (جدول3). فزون بر این، چند شکلیهای حذف و اضافه با FDR کمتر از 1 درصد، بیشتر در مسیرهای بیولوژیک مرتبط با تنظیم فرآیند رونویسی، متابولیسم فسفر، چسبندگی سلولی، حرکت سلولی، تمایز نرونها، سازمانبندی اسکلت سلولی، تکامل جنینی، چرخه سلولی، متابولیسم ماکرومولکولها، تمایز رگی، تمایز اندام جنینی، فرآیندهای مرتبط با فیلامنتهای حدواسط، اندوسیتوز، تنظیم حرکت و مهاجرت سلول درگیر هستند. فزون بر چندشکلیهای تک نوکلیوتیدی نامعنی و حذف و اضافههای کوچک، آنالیز
Gene Ontology برای ژنهای دارای واریانتهای با آثار بالا بر کنش و ساختار ژن نشان داد که این ژنها در مسیرهای بیولوژیک مرتبط با پاسخ ایمنی و انتقال یونی نقش دارند (شکل3).
بحث
توالییابی و مونتاژ ژنوم اسب یکی از دستاوردهای مهم است که کاربرد گستردهای در بهبود عملکرد و سلامت حیوان و نیز درک بیشتر تفاوتهای تکاملی و مولکولی با دیگر پستانداران دارد. تاکنون ژنوم اسب نژادهای Thoroughbred (Wade et al., 2009) و Quarter (Doan et al., 2013) عرب (Orlando et al., 2013)، Icelandic (Orlando et al., 2013)، Standardbred (Orlando et al., 2013)، Przewalskii (Orlando et al., 2013) و Norwegian Fjord (Orlando et al., 2013) توالییابی شده و در دسترساند. شمار کل واریاسیونهای ژنتیکی که تاکنون در اسب شناسایی شدهاند نزدیک 3 میلیون SNP است؛ که از میان بیشتر واریاسیونهای شناخته شده اسب (64 درصد) از نریان Thoroughbred توالییابی و مونتاژ شده به دست آمده است(Doan et al., 2012). با وجود اینکه گفته میشود اسب کاسپین از کهنترین اسبهای اهلی دنیاست و جد اسبهای اورینتال خاورمیانه است ولی پژوهشی برای شناخت ساختار ژنوم و نیز واریانتهای ژنومیک این اسبها انجام نگرفته بود. پژوهش حاضر نخستین تلاش برای شناسایی ژنومیک اسب کاسپین با استفاده از تکنیکهای نوین توالییابی با کارایی بالا است که توانسته است نزدیک 72 درصد از ژنوم اسب کاسپین و واریانتهای آن را رونمایی کند. در ابتدای امر، با وجود عمق توالییابی و شمار زیادی توالی کوتاه که در فرآیند توالییابی اسب کاسپین به دست آمد پیشبینی میشد که بخش قابل توجهی از ژنوم اسب کاسپین پوشش داده شود ولی پس از نقشه یابی توالیهای کوتاه با ژنوم رفرانس اسب دیده شد که 558507789 نوکلیوتید از 2367053447 نوکلیوتید ژنوم هیچ همپوشانی با توالیهای کوتاه ندارند. از این رو، بررسی بیشتر ساختار ژنوم اسب کاسپین با استفاده از De novo Assembly توالیهای کوتاه نقشهیابی نشده با ژنوم رفرانس بایسته مینماید.
هرچند در پژوهشهای مستقل (Doan et al., 2012) و (Orlando et al., 2013) شمار چندشکلیهای شناسایی شده در هر نژاد از اسبها بیش از 3 میلیون بوده است ولی در این پژوهش شمار چندشکلیهای شناسایی شده در ژنوم اسب کاسپین روی هم رفته 1666717 بود. مهمترین علت این امر استفاده از الگوریتمهای با صحت بالاتر و فیلترینگ بسیار شدیدتر روی شناسایی واریانتها بوده است. علت دیگری که احتمالا در بروز تفاوت در شمار واریانتهای شناسایی شده در اسب کاسپین با دیگر ژنومهای اسب در پژوهشهای پیشین موثر بوده، ساختار منحصر به فرد ژنوم اسب کاسپین است. بیشک، بررسیهای بیشتر دادههای این پژوهش با الگوریتمهای فایلوژنتیک ژنومیک پرده از روابط فایلوژنتیک این اسب با دیگر نژادهای اسب ارایه خواهد کرد.
نتایج کلاسترینگ عملکردی غنی بودن واریانتها در ژنهای مربوط به توسعه نورولوژیک و نیز ادراک حسی در اسب کاسپین را به روشنی نشان داد. شاید بتوان خوی آرام و هوش این اسبان را به تنوع ژنتیکی در این دسته از ژنها ارتباط داد؛ هرچند که این بیان بیشتر پایه حدس و گمان دارد و برای روشن شدن نقش دقیقتر واریانت این ژنها در فنوتیپ ذکر شده به بررسیهای ژنتیک جمعیتی با استفاده از ریزآرایههای DNA در جمعیت اسبان کاسپین در آینده نیاز است.
از دیگر دستآوردهای این پژوهش، شناسایی واریانتهای ساختاری ژنوم اسب کاسپین است که با توجه با اهمیتی که این واریانتها در طراحی نشانگرهای ژنومیک در بررسیهای ژنتیک و کارهای اصلاحی در این نژاد کهن اسب ایرانی دارد، امید است که پژوهشهای آینده به این نژاد در حال انقراض توجهی ویژه (با رویکرد ژنتیکی نوین) پیدا کنند.
سپاسگزاری
این پژوهش با حمایت مالی آکادمی علوم چین انجام شده است. فزون بر این، آنالیزهای ژنومیک و دسترسی به منابع موردنیاز در اجرای این پژوهش با همکاری پژوهشکده رویان صورت پذیرفته است. از این رو، مجریان و همکاران مراتب سپاس و قدردانی خود را از این دو پژوهشگاه اعلام میدارند.
منابع
Abyzov A, Urban AE, Snyder M, Gerstein M (2011). CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Research 21: 974-984.
Andrews S (2012). FASTQC. A quality control tool for high throughput sequence data. URL http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP (2009). BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nature Methods 6: 677-681.
Cingolani P, Platts A, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6: 80-92.
Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, Laub T, Nunn G, Stephan DA (2008). Identification of genetic variants using bar-coded multiplexed sequencing. Nature Methods 5: 887-893.
Cronn R, Liston A, Parks M, Gernandt DS, Shen R, Mockler T (2008). Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Research 36: e122-e122.
Dennis Jr G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome Biology 4: P3.
Doan R, Cohen ND, Sawyer J, Ghaffari N, Johnson CD, Dindot SV (2012). Whole-Genome sequencing and genetic variant analysis of a quarter Horse mare. BMC Genomics 13: 78.
Firouz L (1969). Conservation of a domestic breed. Biological Conservation 2: 53-54.
Firouz L (1971). Osteological and historical implication of the Caspian pony to early domestication in Iran. Proc 3rd Int Congr Agricultural Museum, Budapest: 1-5.
Firouz L (1972). The Caspian miniature horse of Iran. Field Research Projects, Florida, USA,
Garrison E, Marth G (2012). Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv:12073907.
Hatami-Monazah H, Pandit RV (1979). A cytogenetic study of the Caspian pony. Journal of Reproduction and Fertility 57: 331-333.
Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan J, Fan D, Weng Q, Huang T (2009). High-throughput genotyping by whole-genome resequencing. Genome Research 19: 1068-1076.
Langmead B (2002). Aligning Short Sequencing Reads with Bowtie. Current Protocols in Bioinformatics: John Wiley & Sons, Inc.
Lindgreen S (2012). AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Research Notes 5: 337.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20: 1297-1303.
Orlando L, Ginolhac Al, Zhang G, Froese D, Albrechtsen A, Stiller M, Schubert M, Cappellini E, Petersen B, Moltke I (2013). Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499: 74-81
Shahsavarani H, Rahimi-Mianji G (2012). Analysis of genetic diversity and estimation of inbreeding coefficient within Caspian horse population using microsatellite markers. African Journal of Biotechnology 9: 293-299
Shao H, Bellos E, Yin H, Liu X, Zou J, Li Y, Wang J, Coin LJM (2012). A population model for genotyping indels from next-generation sequence data. Nucleic acids research 41: e46-e46.
Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, Lear TL, Adelson DL, Bailey E, Bellone RR, Blöcker H, Distl O, Edgar RC, Garber M, Leeb T, Mauceli E, MacLeod JN, Penedo MCT, Raison JM, Sharpe T, Vogel J, Andersson L, Antczak DF, Biagi T, Binns MM, Chowdhary BP, Coleman SJ, Della Valle G, Fryc S, Guérin G, Hasegawa T, Hill EW, Jurka J, Kiialainen A, Lindgren G, Liu J, Magnani E, Mickelson JR, Murray J, Nergadze SG, Onofrio R, Pedroni S, Piras MF, Raudsepp T, Rocchi M, Røed KH, Ryder OA, Searle S, Skow L, Swinburne JE, Syvänen AC, Tozaki T, Valberg SJ, Vaudin M, White JR, Zody MC, Broad Institute Genome Sequencing P, Broad Institute Whole Genome Assembly T, Lander ES, Lindblad-Toh K (2009). Genome Sequence, Comparative Analysis, and Population Genetics of the Domestic Horse. Science 326: 865-867.
Wang K, Li M, Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research 38: e164-e164.
Winzeler EA, Richards DR, Conway AR, Goldstein AL, Kalman S, McCullough MJ, McCusker JH, Stevens DA, Wodicka L, Lockhart DJ (1998). Direct allelic variation scanning of the yeast genome. Science 281: 1194-1197.